

Aleksandra Zalewska-Stankiewicz, „Wprost”: Kim są „dzieci-motyle”?
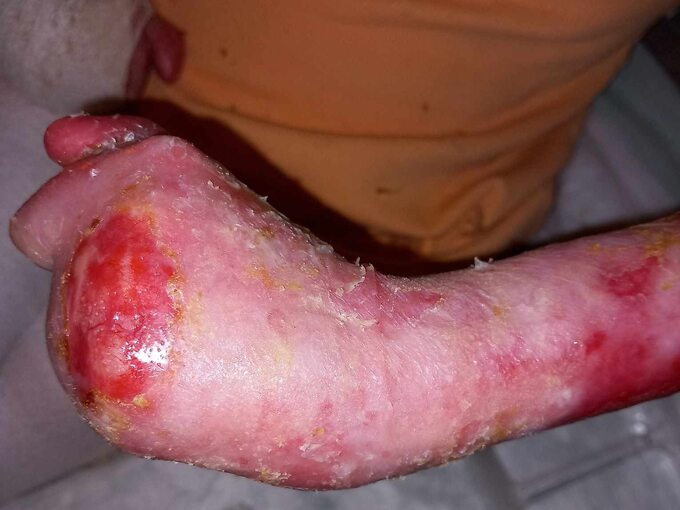
Dr n. med. Katarzyna Wertheim-Tysarowska*: To mali pacjenci dotknięci rzadką nieuleczalną chorobą genetyczną – pęcherzowym oddzielaniem się naskórka, Epidermolysis Bullosa, w skrócie nazywaną EB. Ich skóra faktycznie jest tak krucha i delikatna jak skrzydła motyla. W obrębie ich skóry i błon śluzowych jest mnóstwo pęcherzy i nadżerek. Powstają samoistnie albo wywołuje je nawet najmniejszy uraz, dotyk lub uścisk. Konsekwencją są trudno gojące się rany.
Na stronie Fundacji EB widziałam film, na którym 14-letnia Zosia opowiada o swojej chorobie. Widać, że pacjenci bardzo cierpią.
Ich życie polega na wymianie opatrunków – może trwać nawet kilka godzin dziennie. Pielęgnacja ran to prawdziwe wyzwanie – muszą być dezynfekowane, przekłute igłą i zabezpieczone specjalnymi opatrunkami. Zwykłe plastry podrażniłyby skórę pacjentów, a nawet ją oderwały. Od tej choroby nie ma oddechu, kiedy jedna rana się zagoi, tworzy się kolejna. Pęcherze nie pojawiają się wyłącznie na skórze, mogą wystąpić w jamie ustnej (na języku czy podniebieniu), w oku, a także w przełyku doprowadzają do jego zwężania. Są bardzo bolesne. Mogą prowadzić też do poważnych konsekwencji, jak anemia (pacjenci nie mogą przełykać), deformacje kończyn, przykurcze mięśni, ale i depresje czy zaburzenia lękowe.
Jak to się stało, że zainteresowała się Pani EB?
W 2006 roku rozpoczęłam pracę w Zakładzie Genetyki Medycznej Instytutu Matki i Dziecka (ZGM IMiD). Wówczas nie zajmowaliśmy się tematami skórnymi, a EB zaliczana jest do chorób dermatologicznych. Szukałam tematu do doktoratu, a pacjenci z EB w ogóle nie byli wówczas genetycznie zdiagnozowani. Dr Agnieszka Sobczyńska-Tomaszewska z naszego Zakładu i prof. Cezary Kowalewski z Warszawskiego Uniwersytetu Medycznego postanowili to zmienić. Dzięki temu rozpoczął się projekt badawczy dotyczący genetyki EB, w ramach którego zrobiłam doktorat. Na początku badaliśmy tylko jeden typ EB.
To jest ich więcej?
Tak, wyróżnia się 4 typy EB, w zależności od miejsca powstawania pęcherza na granicy skórno- naskórkowej. Są to: typ prosty, łączący, dystroficzny i zespół Kindlera. Ponadto – w zależności od objawów klinicznych i sposobu dziedziczenia – występuje około 30 podtypów EB. Poszerzaliśmy wiedzę, a jednocześnie zaczęli zgłaszać się do nas pacjenci z całej Polski. W ten sposób w naszym Zakładzie zaczęliśmy również prowadzić diagnostykę molekularną EB. Do chwili obecnej w IMiD zdiagnozowanych jest ponad 170 rodzin z różnymi postaciami EB. W Polsce jest ich więcej, między 200 a 400 rodzin, ale dokładnych danych nie ma.
Jak dziedziczy się EB?
EB jest chorobą genetycznie uwarunkowaną, która może dziedziczyć się w sposób autosomalny recesywny lub dominujący. W pierwszym przypadku oznacza to, że choroba pojawia się wtedy, gdy mutacje występujące w obu kopiach danego genu są odziedziczone od obojga rodziców. W takiej sytuacji rodzice są jedynie nosicielami mutacji i nie ma u nich objawów choroby, natomiast mają 25% ryzyko posiadania dziecka chorego na EB, w przypadku każdej ciąży. W drugim typie dziedziczenia – autosomalnym dominującym – wystarczy mutacja tylko w jednej kopii genu, aby choroba rozwinęła się. Ryzyko przekazania mutacji, a więc pojawienia się choroby u dziecka, wynosi w tym przypadku 50%. Mutacja z reguły występuje de novo.
Jakiego odkrycia dokonaliście podczas badań?
To, że EB jest chorobą uwarunkowaną genetycznie wiedzieliśmy wcześniej. Znanych jest kilkanaście białek, których uszkodzenie (wskutek mutacji w kodujących je genach) powoduje rozwój EB. Każde z tych białek kodowane jest przez inny gen. Podczas realizacji projektu określiliśmy podłoże molekularne, zidentyfikowaliśmy patogenne mutacje w badanych rodzinach, wypracowaliśmy algorytm diagnostyczny, dzięki któremu można było przeprowadzać diagnostykę pacjentów. Proszę pamiętać, że 18 lat temu w Polsce ta grupa chorych nie miała w ogóle diagnozy genetycznej, a badania genetyczne w kierunku EB nie były wykonywane, nie było więc molekularnej weryfikacji rozpoznania klinicznego.
Dzięki nowym technologiom analizy DNA możemy obecnie równocześnie analizować wszystkie znane geny, których mutacje odpowiadają za wystąpienie EB. Nie tak dawno nie było dostępnych badań wielkoskalowych, a każdy gen badało się indywidualnie. Były to badania trudne i pracochłonne ze względu na wielkość genów. Przykładowo analiza genu COL7A1, odpowiedzialnego za tzw. typ dystroficzny EB wymagała zanalizowania aż… 118 fragmentów genu. Zidentyfikowaliśmy m.in. warianty genetyczne, odpowiedzialne za wystąpienie EB występujące najczęściej w naszej polskiej populacji. Z diagnostycznego punktu widzenia ważne były także badania nosicielstwa EB. Dzięki temu można było udzielić porady genetycznej i rozwiać wiele wątpliwości.
Jakie to najczęściej wątpliwości?
W jaki sposób dziedziczy się chorobę, jak duże jest ryzyko powtórzenia się choroby w rodzinie. Czasem, po przeprowadzeniu badań genetycznych, okazuje się, że ryzyko to jest znacznie mniejsze niż zakładane i wynosi jedynie 1-5%. W przypadku niektórych mutacji można przewidzieć przebieg kliniczny choroby. Z naukowego punktu widzenia najciekawsi okazali się pacjenci z klinicznym rozpoznaniem EB, u których nie zidentyfikowaliśmy żadnych mutacji. Mamy nadzieję, że kontynuując badania, opiszemy mutacje i geny, w których występują, a które do tej pory nie były kojarzone z występowaniem EB. Pamiętajmy, że lista genów odpowiedzialnych za tę chorobę nie jest zamknięta. W naskórku swoja ekspresje ma tysiące genów, a funkcji wielu z nich jeszcze nie poznaliśmy. Warto też wspomnieć, że na podstawie badań genetycznych można zakwalifikować pacjentów do programów terapeutycznych i badań klinicznych. W chwili obecnej jest to w zasadzie jedno z kryteriów włączenia.
Gdy projekt się zakończył, Pani nadal próbowała odkrywać kolejne sekrety EB. Co Panią kierowało ?
Choć nie jestem lekarzem, lecz genetykiem, widziałam ogromne cierpienie pacjentów, którzy zmagali się z EB przez całe życie. U niektórych ta choroba przebiega łagodnie, ale jest grupa osób, które przeżywają prawdziwe dramaty. Ich skóra jest zajęta zmianami w 80 procentach, muszą być cały czas bandażowane. Ta choroba wymaga opieki wielospecjalistycznej – dermatologa, pediatry, chirurga, stomatologa, otolaryngologa, okulisty, kardiologa, nefrologa, dietetyka, rehabilitanta, psychoterapeuty. Dostrzegłam, że brakuje specjalistów specjalizujących się w leczeniu EB. Za granicą było inaczej.
To znaczy jak?
Pojechałam na miesiąc do Salzburga, gdzie znajduje się EB House, ośrodek specjalizujący się w EB. Zobaczyłam, jak może wyglądać wielospecjalistyczna opieka nad pacjentem z EB. Mogą skonsultować z lekarzami każdy problem, nie tylko ten skórny, ale też ze słuchem, wzrokiem, gardłem czy uzębieniem. Tymczasem w Polsce brakowało i nadal niestety brakuje lekarzy wielu specjalności doświadczonych w EB. W efekcie wielu z nich boi się badać pacjentów dotkniętych tą chorobą, ze względu na ryzyko uszkodzenia skóry czy błon śluzowych. Ten problem dostrzegli rodzice dzieci z EB. To dzięki nim powstała Fundacja EB Polska. Również byłam w gronie założycielskim.
Czym zajmuje się Fundacja EB?
Na początku nawiązaliśmy współpracę z dr Marcinem Malką z Kliniki Leczenia Ran PODOS. Zaczęliśmy organizować szkolenia dla pacjentów, głównie dotyczące pielęgnacji ran. Dużym zainteresowaniem cieszyły się szkolenia dla personelu medycznego – nasz fundacyjny zespół jechał do szpitala, w którym urodziło się dziecko z EB i w praktyce pokazywał, jak powinna wyglądać opieka. Dodatkowo przekazywana jest tzw. pierwsza paczka dla małych pacjentów, pełna specjalistycznych opatrunków. Jak widać, w moim przypadku zaczęło się od badań genetycznych, a skończyło na rozwoju społeczności skupionej wokół EB. Choć wciąż przed nami wiele wyzwań – w Polsce brakuje systemowej opieki dla pacjentów z tą chorobą.
Jednak badania genetyczne dały początek pozytywnym zmianom. Jakim?
Rzeczywiście, kilkanaście lat temu mieliśmy pacjentów jedynie z klinicznym rozpoznaniem EB, o genetyce nie wiedzieliśmy wtedy nic. Kamieniem milowym było wprowadzenie badań genetycznych i opracowanie schematu diagnostyki dla chorych na EB, co też zostało docenione za granicą. Zostałam zaproszona – jako jedna z kilkorga specjalistów z całego świata – do opracowania międzynarodowych wytycznych.
Jestem dumna, że razem z zespołem ZGM IMiD udało się nam poszerzyć wiedzę na temat zależności między obrazem klinicznym a genotypem pacjenta, co ma bezpośrednie przełożenie na poradnictwo genetyczne. W tej chwili genetyka jest standardem w diagnozowaniu EB. Dzięki działaniom m.in. Fundacji EB Polska pojawiły się wytyczne neonatologiczne dla pacjentów. Sukcesem, do którego się również przyczyniliśmy, było wywalczenia świadczenia opatrunkowego dla chorych, którzy używają ogromnych ilości materiałów do pielęgnacji, a to jest bardzo kosztowne.
Jakie wyzwania w tej materii stoją przed genetykami?
Wiedza o patologii molekularnej EB przyczyni się do wypracowania nowych metod leczenia, w tym leczenia celowanego. Przymierzamy się do projektu naukowego, początkowo na modelach komórkowych, którego celem będzie eliminowanie patogennych mutacji w genach odpowiedzialnych za EB. Kierunek wyznaczają jednak badania w USA, gdzie opracowano terapię genową EB. W terapii genowej podstawowym problemem jest docelowe dostarczenie zmodyfikowanego preparatu. W przypadku EB problem ten rozwiązano terapię genową w postaci żelu na rany. Prawdopodobnie ten typ leczenia w przyszłym roku będzie dostępny w Europie. W tym roku pojawił się natomiast na rynku pierwszy lek zarejestrowany dla EB, więc też jest to kolejny krok w stronę opracowania leczenia tej okrutnej choroby. W Polsce jest on na razie dostępny niestety wyłącznie w trybie ratunkowego dostępu do technologii lekowych. Ale pierwsza Polka otrzymała już ten lek – to Zosia, o której Pani wspominała.
Jednak największym wyzwaniem, i to nie genetycznym, jest zagwarantowanie polskim chorym należytej opieki. Chorzy na EB muszą mieć zapewniony dostęp do specjalistycznego ośrodka, powinno zostać powołane – wzorem innych krajów – stanowisko pielęgniarki EB. Natomiast rodzice chorych dzieci muszą mieć zapewnione profesjonalne wsparcie, w tym w ramach opieki wytchnieniowej.
*Dr n. med. Katarzyna Wertheim-Tysarowska, Zakład Genetyki Medycznej, Instytut Matki i Dziecka, Pracownia Badań Chorób Dziedzicznych, specjalista w zakresie laboratoryjnej genetyki medycznej, autorka i współautorka ponad 50 artykułów z zakresu genetyki człowieka oraz rozdziałów książkowych, laureatka nagrody Prezesa Rady Ministrów za pracę doktorską oraz nagród Polskiego Towarzystwa Genetyk za pracę doktorską i publikacje. Zainteresowania badawcze koncentrują się wokół etiopatogenezy molekularnej chorób uwarunkowanych genetycznie, w szczególności chorób skóry, chorób metabolicznych i niepłodności.
Nauka to polska specjalność
Wielkie postacie polskiej nauki
Przeczytaj inne artykuły poświęcone polskiej nauce
Projekt współfinansowany ze środków Ministerstwa Edukacji i Nauki w ramach programu „Społeczna Odpowiedzialność Nauki”